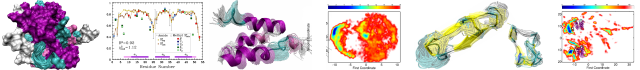

Journal Articles
A large-scale conformation sampling and evaluation server for protein tertiary structure prediction and its assessment in CASP11
Phylogenomics and sequence-structure-function relationships in the GmrSD family of Type IV restriction enzymes
AlloPred: prediction of allosteric pockets on proteins using normal mode perturbation analysis
Intracellular Information Processing through Encoding and Decoding of Dynamic Signaling Features
by Hirenkumar K. Makadia, James S. Schwaber, Rajanikanth Vadigepalli
Cell signaling dynamics and transcriptional regulatory activities are variable within specific cell types responding to an identical stimulus. In addition to studying the network interactions, there is much interest in utilizing single cell scale data to elucidate the non-random aspects of the variability involved in cellular decision making. Previous studies have considered the information transfer between the signaling and transcriptional domains based on an instantaneous relationship between the molecular activities. These studies predict a limited binary on/off encoding mechanism which underestimates the complexity of biological information processing, and hence the utility of single cell resolution data. Here we pursue a novel strategy that reformulates the information transfer problem as involving dynamic features of signaling rather than molecular abundances. We pursue a computational approach to test if and how the transcriptional regulatory activity patterns can be informative of the temporal history of signaling. Our analysis reveals (1) the dynamic features of signaling that significantly alter transcriptional regulatory patterns (encoding), and (2) the temporal history of signaling that can be inferred from single cell scale snapshots of transcriptional activity (decoding). Immediate early gene expression patterns were informative of signaling peak retention kinetics, whereas transcription factor activity patterns were informative of activation and deactivation kinetics of signaling. Moreover, the information processing aspects varied across the network, with each component encoding a selective subset of the dynamic signaling features. We developed novel sensitivity and information transfer maps to unravel the dynamic multiplexing of signaling features at each of these network components. Unsupervised clustering of the maps revealed two groups that aligned with network motifs distinguished by transcriptional feedforward vs feedback interactions. Our new computational methodology impacts the single cell scale experiments by identifying downstream snapshot measures required for inferring specific dynamical features of upstream signals involved in the regulation of cellular responses.Ten Simple Rules for Creating a Good Data Management Plan
by William K. Michener
Laminar Neural Field Model of Laterally Propagating Waves of Orientation Selectivity
by Paul C. Bressloff, Samuel R. Carroll
We construct a laminar neural-field model of primary visual cortex (V1) consisting of a superficial layer of neurons that encode the spatial location and orientation of a local visual stimulus coupled to a deep layer of neurons that only encode spatial location. The spatially-structured connections in the deep layer support the propagation of a traveling front, which then drives propagating orientation-dependent activity in the superficial layer. Using a combination of mathematical analysis and numerical simulations, we establish that the existence of a coherent orientation-selective wave relies on the presence of weak, long-range connections in the superficial layer that couple cells of similar orientation preference. Moreover, the wave persists in the presence of feedback from the superficial layer to the deep layer. Our results are consistent with recent experimental studies that indicate that deep and superficial layers work in tandem to determine the patterns of cortical activity observed in vivo.Qualitative and Quantitative Protein Complex Prediction Through Proteome-Wide Simulations
by Simone Rizzetto, Corrado Priami, Attila Csikász-Nagy
Despite recent progress in proteomics most protein complexes are still unknown. Identification of these complexes will help us understand cellular regulatory mechanisms and support development of new drugs. Therefore it is really important to establish detailed information about the composition and the abundance of protein complexes but existing algorithms can only give qualitative predictions. Herein, we propose a new approach based on stochastic simulations of protein complex formation that integrates multi-source data—such as protein abundances, domain-domain interactions and functional annotations—to predict alternative forms of protein complexes together with their abundances. This method, called SiComPre (Simulation based Complex Prediction), achieves better qualitative prediction of yeast and human protein complexes than existing methods and is the first to predict protein complex abundances. Furthermore, we show that SiComPre can be used to predict complexome changes upon drug treatment with the example of bortezomib. SiComPre is the first method to produce quantitative predictions on the abundance of molecular complexes while performing the best qualitative predictions. With new data on tissue specific protein complexes becoming available SiComPre will be able to predict qualitative and quantitative differences in the complexome in various tissue types and under various conditions.Ion-Exchangeable Molybdenum Sulfide Porous Chalcogel: Gas Adsorption and Capture of Iodine and Mercury
Organic Radical-Assisted Electrochemical Exfoliation for the Scalable Production of High-Quality Graphene
N-Acyl Amino Acid Ligands for Ruthenium(II)-Catalyzed meta-C–H tert-Alkylation with Removable Auxiliaries
Sialic Acid-Imprinted Fluorescent Core–Shell Particles for Selective Labeling of Cell Surface Glycans
Photoinduced, Copper-Catalyzed Carbon–Carbon Bond Formation with Alkyl Electrophiles: Cyanation of Unactivated Secondary Alkyl Chlorides at Room Temperature
A Cu/Pd Cooperative Catalysis for Enantioselective Allylboration of Alkenes
Facile Conversion of Hydroxy Double Salts to Metal–Organic Frameworks Using Metal Oxide Particles and Atomic Layer Deposition Thin-Film Templates
Organometallic Complexes Anchored to Conductive Carbon for Electrocatalytic Oxidation of Methane at Low Temperature
Elucidating nitric oxide synthase domain interactions by molecular dynamics
Nitric oxide synthase (NOS) is a multidomain enzyme that catalyzes the production of nitric oxide (NO) by oxidizing l-Arg to NO and L-citrulline. NO production requires multiple interdomain electron transfer steps between the flavin mononucleotide (FMN) and heme domain. Specifically, NADPH-derived electrons are transferred to the heme-containing oxygenase domain via the flavin adenine dinucleotide (FAD) and FMN containing reductase domains. While crystal structures are available for both the reductase and oxygenase domains of NOS, to date there is no atomic level structural information on domain interactions required for the final FMN-to-heme electron transfer step. Here, we evaluate a model of this final electron transfer step for the heme–FMN–calmodulin NOS complex based on the recent biophysical studies using a 105-ns molecular dynamics trajectory. The resulting equilibrated complex structure is very stable and provides a detailed prediction of interdomain contacts required for stabilizing the NOS output state. The resulting equilibrated complex model agrees well with previous experimental work and provides a detailed working model of the final NOS electron transfer step required for NO biosynthesis.
The origin of β-strand bending in globular proteins
Path Similarity Analysis: A Method for Quantifying Macromolecular Pathways
by Sean L. Seyler, Avishek Kumar, M. F. Thorpe, Oliver Beckstein
Diverse classes of proteins function through large-scale conformational changes and various sophisticated computational algorithms have been proposed to enhance sampling of these macromolecular transition paths. Because such paths are curves in a high-dimensional space, it has been difficult to quantitatively compare multiple paths, a necessary prerequisite to, for instance, assess the quality of different algorithms. We introduce a method named Path Similarity Analysis (PSA) that enables us to quantify the similarity between two arbitrary paths and extract the atomic-scale determinants responsible for their differences. PSA utilizes the full information available in 3N-dimensional configuration space trajectories by employing the Hausdorff or Fréchet metrics (adopted from computational geometry) to quantify the degree of similarity between piecewise-linear curves. It thus completely avoids relying on projections into low dimensional spaces, as used in traditional approaches. To elucidate the principles of PSA, we quantified the effect of path roughness induced by thermal fluctuations using a toy model system. Using, as an example, the closed-to-open transitions of the enzyme adenylate kinase (AdK) in its substrate-free form, we compared a range of protein transition path-generating algorithms. Molecular dynamics-based dynamic importance sampling (DIMS) MD and targeted MD (TMD) and the purely geometric FRODA (Framework Rigidity Optimized Dynamics Algorithm) were tested along with seven other methods publicly available on servers, including several based on the popular elastic network model (ENM). PSA with clustering revealed that paths produced by a given method are more similar to each other than to those from another method and, for instance, that the ENM-based methods produced relatively similar paths. PSA applied to ensembles of DIMS MD and FRODA trajectories of the conformational transition of diphtheria toxin, a particularly challenging example, showed that the geometry-based FRODA occasionally sampled the pathway space of force field-based DIMS MD. For the AdK transition, the new concept of a Hausdorff-pair map enabled us to extract the molecular structural determinants responsible for differences in pathways, namely a set of conserved salt bridges whose charge-charge interactions are fully modelled in DIMS MD but not in FRODA. PSA has the potential to enhance our understanding of transition path sampling methods, validate them, and to provide a new approach to analyzing conformational transitions.Crawling and Gliding: A Computational Model for Shape-Driven Cell Migration
by Ioana Niculescu, Johannes Textor, Rob J. de Boer
Cell migration is a complex process involving many intracellular and extracellular factors, with different cell types adopting sometimes strikingly different morphologies. Modeling realistically behaving cells in tissues is computationally challenging because it implies dealing with multiple levels of complexity. We extend the Cellular Potts Model with an actin-inspired feedback mechanism that allows small stochastic cell rufflings to expand to cell protrusions. This simple phenomenological model produces realistically crawling and deforming amoeboid cells, and gliding half-moon shaped keratocyte-like cells. Both cell types can migrate randomly or follow directional cues. They can squeeze in between other cells in densely populated environments or migrate collectively. The model is computationally light, which allows the study of large, dense and heterogeneous tissues containing cells with realistic shapes and migratory properties.