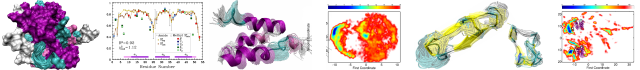

Journal of Structural Biology
M-free: Mask-independent scoring of the reference bias
Publication date: Available online 29 August 2015
Source:Journal of Structural Biology
Author(s): Michael Kunz, Zhou Yu, Achilleas S. Frangakis
The M-free score is a heuristic to measure the reference bias in applications such as template matching and sub-tomogram averaging. In the original formulation the mask typically used in these applications had to be separated into a working and a testing area. Here we present a variant of the calculation of the M-free score, which under certain conditions does not require adapting the mask used during the processing. This is made possible by a modified algorithm that allows for arbitrary variances in the testing and in the working area. Consequently, the reference bias can be estimated with knowledge of only the starting reference, the final average and the mask used for processing. We show that the new formulation of the M-free score gives a reliable measure of the reference bias for any sub-tomogram average that has ancillary data, such as when the averaged structure contains density in the periphery, when a complex is attached to a membrane (membrane-associated complexes) or when one subunit is attached to others (e.g. in viruses). Further, we show that in contrast to correlation-based measurements, the M-free score is sensitive to wrong-alignments and contaminations present in the data set. The scope of this new calculation of the M-free score is to reduce the constraints of the previous approach and in certain cases to avoid an adaptation of the mask. The M-free score gives a separate reliability measure for sub-tomogram averaging and template matching.
Source:Journal of Structural Biology
Author(s): Michael Kunz, Zhou Yu, Achilleas S. Frangakis
The M-free score is a heuristic to measure the reference bias in applications such as template matching and sub-tomogram averaging. In the original formulation the mask typically used in these applications had to be separated into a working and a testing area. Here we present a variant of the calculation of the M-free score, which under certain conditions does not require adapting the mask used during the processing. This is made possible by a modified algorithm that allows for arbitrary variances in the testing and in the working area. Consequently, the reference bias can be estimated with knowledge of only the starting reference, the final average and the mask used for processing. We show that the new formulation of the M-free score gives a reliable measure of the reference bias for any sub-tomogram average that has ancillary data, such as when the averaged structure contains density in the periphery, when a complex is attached to a membrane (membrane-associated complexes) or when one subunit is attached to others (e.g. in viruses). Further, we show that in contrast to correlation-based measurements, the M-free score is sensitive to wrong-alignments and contaminations present in the data set. The scope of this new calculation of the M-free score is to reduce the constraints of the previous approach and in certain cases to avoid an adaptation of the mask. The M-free score gives a separate reliability measure for sub-tomogram averaging and template matching.
Categories: Journal Articles
Structural characterization and modeling of the Borrelia burgdorferi hybrid histidine kinase Hk1 periplasmic sensor: A system for sensing small molecules associated with tick feeding
Publication date: Available online 28 August 2015
Source:Journal of Structural Biology
Author(s): William J. Bauer, Amit Luthra, Guangyu Zhu, Justin D. Radolf, Michael G. Malkowski, Melissa J. Caimano
Two-component signal transduction systems are the primary mechanisms by which bacteria perceive and respond to changes in their environment. The Hk1/Rrp1 two-component system (TCS) in Borrelia burgdorferi consists of a hybrid histidine kinase and a response regulator with diguanylate cyclase activity, respectively. Phosphorylated Rrp1 catalyzes the synthesis of c-di-GMP, a second messenger associated with bacterial life-style control networks. Spirochetes lacking either Hk1 or Rrp1 are virulent in mice but destroyed within feeding ticks. Activation of Hk1 by exogenous stimuli represents the seminal event for c-di-GMP signaling. We reasoned that structural characterization of Hk1’s sensor would provide insights into the mechanism underlying signal transduction and aid in the identification of activating ligands. The Hk1 sensor is composed of three ligand-binding domains (D1–3), each with homology to periplasmic solute-binding proteins (PBPs) typically associated with ABC transporters. Herein, we determined the structure for D1, the most N-terminal PBP domain. As expected, D1 displays a bilobed Venus Fly Trap-fold. Similar to the prototypical sensor PBPs HK29S from Geobacter sulfurreducens and VFT2 from Bordetella pertussis, apo-D1 adopts a closed conformation. Using complementary approaches, including SAXS, we established that D1 forms a dimer in solution. The D1 structure enabled us to model the D2 and D3 domains. Differences in the ligand-binding pockets suggest that each PBP recognizes a different ligand. The ability of Hk1 to recognize multiple stimuli provides spirochetes with a means of distinguishing between the acquisition and transmission blood meals and generate a graded output response that is reflective of the perceived environmental threats.
Source:Journal of Structural Biology
Author(s): William J. Bauer, Amit Luthra, Guangyu Zhu, Justin D. Radolf, Michael G. Malkowski, Melissa J. Caimano
Two-component signal transduction systems are the primary mechanisms by which bacteria perceive and respond to changes in their environment. The Hk1/Rrp1 two-component system (TCS) in Borrelia burgdorferi consists of a hybrid histidine kinase and a response regulator with diguanylate cyclase activity, respectively. Phosphorylated Rrp1 catalyzes the synthesis of c-di-GMP, a second messenger associated with bacterial life-style control networks. Spirochetes lacking either Hk1 or Rrp1 are virulent in mice but destroyed within feeding ticks. Activation of Hk1 by exogenous stimuli represents the seminal event for c-di-GMP signaling. We reasoned that structural characterization of Hk1’s sensor would provide insights into the mechanism underlying signal transduction and aid in the identification of activating ligands. The Hk1 sensor is composed of three ligand-binding domains (D1–3), each with homology to periplasmic solute-binding proteins (PBPs) typically associated with ABC transporters. Herein, we determined the structure for D1, the most N-terminal PBP domain. As expected, D1 displays a bilobed Venus Fly Trap-fold. Similar to the prototypical sensor PBPs HK29S from Geobacter sulfurreducens and VFT2 from Bordetella pertussis, apo-D1 adopts a closed conformation. Using complementary approaches, including SAXS, we established that D1 forms a dimer in solution. The D1 structure enabled us to model the D2 and D3 domains. Differences in the ligand-binding pockets suggest that each PBP recognizes a different ligand. The ability of Hk1 to recognize multiple stimuli provides spirochetes with a means of distinguishing between the acquisition and transmission blood meals and generate a graded output response that is reflective of the perceived environmental threats.
Categories: Journal Articles
Evaluation of super-resolution performance of the K2 electron-counting camera using 2D crystals of aquaporin-0
Publication date: Available online 28 August 2015
Source:Journal of Structural Biology
Author(s): Po-Lin Chiu, Xueming Li, Zongli Li, Brian Beckett, Axel F. Brilot, Nikolaus Grigorieff, David A. Agard, Yifan Cheng, Thomas Walz
The K2 Summit camera was initially the only commercially available direct electron detection camera that was optimized for high-speed counting of primary electrons and was also the only one that implemented centroiding so that the resolution of the camera can be extended beyond the Nyquist limit set by the physical pixel size. In this study, we used well-characterized two-dimensional crystals of the membrane protein aquaporin-0 to characterize the performance of the camera below and beyond the physical Nyquist limit and to measure the influence of electron dose rate on image amplitudes and phases.
Source:Journal of Structural Biology
Author(s): Po-Lin Chiu, Xueming Li, Zongli Li, Brian Beckett, Axel F. Brilot, Nikolaus Grigorieff, David A. Agard, Yifan Cheng, Thomas Walz
The K2 Summit camera was initially the only commercially available direct electron detection camera that was optimized for high-speed counting of primary electrons and was also the only one that implemented centroiding so that the resolution of the camera can be extended beyond the Nyquist limit set by the physical pixel size. In this study, we used well-characterized two-dimensional crystals of the membrane protein aquaporin-0 to characterize the performance of the camera below and beyond the physical Nyquist limit and to measure the influence of electron dose rate on image amplitudes and phases.
Categories: Journal Articles
X-ray structural and molecular dynamical studies of the globular domains of cow, deer, elk and Syrian hamster prion proteins
Publication date: Available online 28 August 2015
Source:Journal of Structural Biology
Author(s): Pravas Kumar Baral, Mridula Swayampakula, Adriano Aguzzi, Michael N.G. James
Misfolded prion proteins are the cause of neurodegenerative diseases that affect many mammalian species, including humans. Transmission of the prion diseases poses a considerable public-health risk as a specific prion disease such as bovine spongiform encephalopathy can be transferred to humans and other mammalian species upon contaminant exposure. The underlying mechanism of prion propagation and the species barriers that control cross species transmission has been investigated quite extensively. So far a number of prion strains have been characterized and those have been intimately linked to species-specific infectivity and other pathophysiological manifestations. These strains are encoded by a protein-only agent, and have a high degree of sequence identity across mammalian species. The molecular events that lead to strain differentiation remain elusive. In order to contribute to the understanding of strain differentiation, we have determined the crystal structures of the globular, folded domains of four prion proteins (cow, deer, elk and Syrian hamster) bound to the POM1 antibody fragment Fab. Although the overall structural folds of the mammalian prion proteins remains extremely similar, there are several local structural variations observed in the misfolding-initiator motifs. In additional molecular dynamics simulation studies on these several prion proteins reveal differences in the local fluctuations and imply that these differences have possible roles in the unfolding of the globular domains. These local variations in the structured domains perpetuate diverse patterns of prion misfolding and possibly facilitate the strain selection and adaptation.
Source:Journal of Structural Biology
Author(s): Pravas Kumar Baral, Mridula Swayampakula, Adriano Aguzzi, Michael N.G. James
Misfolded prion proteins are the cause of neurodegenerative diseases that affect many mammalian species, including humans. Transmission of the prion diseases poses a considerable public-health risk as a specific prion disease such as bovine spongiform encephalopathy can be transferred to humans and other mammalian species upon contaminant exposure. The underlying mechanism of prion propagation and the species barriers that control cross species transmission has been investigated quite extensively. So far a number of prion strains have been characterized and those have been intimately linked to species-specific infectivity and other pathophysiological manifestations. These strains are encoded by a protein-only agent, and have a high degree of sequence identity across mammalian species. The molecular events that lead to strain differentiation remain elusive. In order to contribute to the understanding of strain differentiation, we have determined the crystal structures of the globular, folded domains of four prion proteins (cow, deer, elk and Syrian hamster) bound to the POM1 antibody fragment Fab. Although the overall structural folds of the mammalian prion proteins remains extremely similar, there are several local structural variations observed in the misfolding-initiator motifs. In additional molecular dynamics simulation studies on these several prion proteins reveal differences in the local fluctuations and imply that these differences have possible roles in the unfolding of the globular domains. These local variations in the structured domains perpetuate diverse patterns of prion misfolding and possibly facilitate the strain selection and adaptation.
Categories: Journal Articles
PAPP-A affects tendon structure and mechanical properties
Publication date: Available online 22 August 2015
Source:Journal of Structural Biology
Author(s): Tai-Hua Yang, Andrew R. Thoreson, Kai-Nan An, Chunfeng Zhao, Cheryl A. Conover, Peter C. Amadio
Pregnancy-associated plasma protein-A (PAPP-A) serves to increase local insulin-like growth factor (IGF) stimulation of proliferation and differentiation in many tissues through proteolysis of inhibitory IGF-binding proteins. The purpose of this study was to investigate the effects of PAPP-A on tendon structure and mechanical properties. A total of 30 tails from 6-month-old mice were tested with 10 tails in each of following groups: PAPP-A knockout (KO), skeletal-specific PAPP-A overexpressing transgenic (Tg) and wild type (WT). Morphologically, the total tail cross-sectional area (CSA), individual tissue CSAs of bone, muscle and tendon, and fascicle diameter were measured. A fascicle pullout test was performed to assess stiffness and strength of interfascicular structures. Fascicles were mechanically characterized through low and high displacement rate uniaxial tension tests providing modulus at each rate, hysteresis area and stress relaxation ratio. The KO mice had a smaller total tail CSA (p <0.05), fascicle diameter (p <0.05), absolute tendon CSA (p <0.05), fast and slow stiffness (p <0.05 for both) and larger hysteresis area (p <0.05) compared to WT and Tg mice. On the other hand, the Tg mice had a larger fascicle diameter (p <0.05), absolute tendon CSA (p <0.05), higher interfascicular strength and stiffness (p <0.05) and lower fascicular modulus at low displacement rates (p <0.05) compared to WT and KO mice. Tg mice also had larger total tail CSA area (p <0.05) and smaller hysteresis area (p <0.05) than KO mice, and larger normalized tendon CSA (p <0.05) than WT mice. Based on these data, we conclude that PAPP-A affects fascicle structure, thereby affecting tendon phenotype.
Source:Journal of Structural Biology
Author(s): Tai-Hua Yang, Andrew R. Thoreson, Kai-Nan An, Chunfeng Zhao, Cheryl A. Conover, Peter C. Amadio
Pregnancy-associated plasma protein-A (PAPP-A) serves to increase local insulin-like growth factor (IGF) stimulation of proliferation and differentiation in many tissues through proteolysis of inhibitory IGF-binding proteins. The purpose of this study was to investigate the effects of PAPP-A on tendon structure and mechanical properties. A total of 30 tails from 6-month-old mice were tested with 10 tails in each of following groups: PAPP-A knockout (KO), skeletal-specific PAPP-A overexpressing transgenic (Tg) and wild type (WT). Morphologically, the total tail cross-sectional area (CSA), individual tissue CSAs of bone, muscle and tendon, and fascicle diameter were measured. A fascicle pullout test was performed to assess stiffness and strength of interfascicular structures. Fascicles were mechanically characterized through low and high displacement rate uniaxial tension tests providing modulus at each rate, hysteresis area and stress relaxation ratio. The KO mice had a smaller total tail CSA (p <0.05), fascicle diameter (p <0.05), absolute tendon CSA (p <0.05), fast and slow stiffness (p <0.05 for both) and larger hysteresis area (p <0.05) compared to WT and Tg mice. On the other hand, the Tg mice had a larger fascicle diameter (p <0.05), absolute tendon CSA (p <0.05), higher interfascicular strength and stiffness (p <0.05) and lower fascicular modulus at low displacement rates (p <0.05) compared to WT and KO mice. Tg mice also had larger total tail CSA area (p <0.05) and smaller hysteresis area (p <0.05) than KO mice, and larger normalized tendon CSA (p <0.05) than WT mice. Based on these data, we conclude that PAPP-A affects fascicle structure, thereby affecting tendon phenotype.
Categories: Journal Articles
Absolute polarity determination of teeth cementum by phase sensitive second harmonic generation microscopy
Publication date: Available online 20 August 2015
Source:Journal of Structural Biology
Author(s): Hanane Aboulfadl, Jürg Hulliger
The absolute sign of local polarity in relation to the biological growth direction has been investigated for teeth cementum using phase sensitive second harmonic generation microscopy (PS-SHGM) and a crystal of 2-cyclooctylamino-5-nitropyridine (COANP) as a nonlinear optic (NLO) reference material. A second harmonic generation (SHG) response was found in two directions of cementum: radial (acellular extrinsic fibers that are oriented more or less perpendicular to the root surface) and circumferential (cellular intrinsic fibers that are oriented more or less parallel to the surface). A mono-polar state was demonstrated for acellular extrinsic cementum. However, along the different parts of cementum in circumferential direction, two corresponding domains were observed featuring an opposite sign of polarity indicative for a bi-polar microscopic state of cellular intrinsic cementum. The phase information showed that the orientation of radial collagen fibrils of cementum is regularly organized with the donor (D) groups pointing to the surface. Circumferential collagen molecules feature orientational disorder and are oriented up and down in random manner showing acceptor or donor groups at the surface of cementum. Considering that the cementum continues to grow in thickness throughout life, we can conclude that the cementum is growing circumferentially in two opposite directions and radially in one direction. A Markov chain type model for polarity formation in the direction of growth predicts D-groups preferably appearing at the fiber front.
Source:Journal of Structural Biology
Author(s): Hanane Aboulfadl, Jürg Hulliger
The absolute sign of local polarity in relation to the biological growth direction has been investigated for teeth cementum using phase sensitive second harmonic generation microscopy (PS-SHGM) and a crystal of 2-cyclooctylamino-5-nitropyridine (COANP) as a nonlinear optic (NLO) reference material. A second harmonic generation (SHG) response was found in two directions of cementum: radial (acellular extrinsic fibers that are oriented more or less perpendicular to the root surface) and circumferential (cellular intrinsic fibers that are oriented more or less parallel to the surface). A mono-polar state was demonstrated for acellular extrinsic cementum. However, along the different parts of cementum in circumferential direction, two corresponding domains were observed featuring an opposite sign of polarity indicative for a bi-polar microscopic state of cellular intrinsic cementum. The phase information showed that the orientation of radial collagen fibrils of cementum is regularly organized with the donor (D) groups pointing to the surface. Circumferential collagen molecules feature orientational disorder and are oriented up and down in random manner showing acceptor or donor groups at the surface of cementum. Considering that the cementum continues to grow in thickness throughout life, we can conclude that the cementum is growing circumferentially in two opposite directions and radially in one direction. A Markov chain type model for polarity formation in the direction of growth predicts D-groups preferably appearing at the fiber front.
Categories: Journal Articles
Structural and computational dissection of the catalytic mechanism of the inorganic pyrophosphatase from Mycobacterium tuberculosis
Publication date: Available online 19 August 2015
Source:Journal of Structural Biology
Author(s): Andrew C. Pratt, Sajeewa W. Dewage, Allan H. Pang, Tapan Biswas, Sandra Barnard-Britson, G. Andrés Cisneros, Oleg V. Tsodikov
Family I inorganic pyrophosphatases (PPiases) are ubiquitous enzymes that are critical for phosphate metabolism in all domains of life. The detailed catalytic mechanism of these enzymes, including the identity of the general base, is not fully understood. We determined a series of crystal structures of the PPiase from Mycobacterium tuberculosis (Mtb PPiase) bound to catalytic metals, inorganic pyrophosphate (PPi; the reaction substrate) and to one or two inorganic phosphate ions (Pi; the reaction product), ranging in resolution from 1.85 to 3.30Å. These structures represent a set of major kinetic intermediates in the catalytic turnover pathway for this enzyme and suggest an order of association and dissociation of the divalent metals, the substrate and the two products during the catalytic turnover. The active site of Mtb PPiase exhibits significant structural differences from the well characterized Escherichia coli PPiase in the vicinity of the bound PPi substrate. Prompted by these differences, quantum mechanics/molecular mechanics (QM/MM) analysis yielded an atomic description of the hydrolysis step for Mtb PPiase and, unexpectedly, indicated that Asp89, rather than Asp54 that was proposed for E. coli PPiase, can abstract a proton from a water molecule to activate it for a nucleophilic attack on the PPi substrate. Mutagenesis studies of the key Asp residues of Mtb PPiase supported this mechanism. This combination of structural and computational analyses clarifies our understanding of the mechanism of family I PPiases and has potential utility for rational development of drugs targeting this enzyme.
Source:Journal of Structural Biology
Author(s): Andrew C. Pratt, Sajeewa W. Dewage, Allan H. Pang, Tapan Biswas, Sandra Barnard-Britson, G. Andrés Cisneros, Oleg V. Tsodikov
Family I inorganic pyrophosphatases (PPiases) are ubiquitous enzymes that are critical for phosphate metabolism in all domains of life. The detailed catalytic mechanism of these enzymes, including the identity of the general base, is not fully understood. We determined a series of crystal structures of the PPiase from Mycobacterium tuberculosis (Mtb PPiase) bound to catalytic metals, inorganic pyrophosphate (PPi; the reaction substrate) and to one or two inorganic phosphate ions (Pi; the reaction product), ranging in resolution from 1.85 to 3.30Å. These structures represent a set of major kinetic intermediates in the catalytic turnover pathway for this enzyme and suggest an order of association and dissociation of the divalent metals, the substrate and the two products during the catalytic turnover. The active site of Mtb PPiase exhibits significant structural differences from the well characterized Escherichia coli PPiase in the vicinity of the bound PPi substrate. Prompted by these differences, quantum mechanics/molecular mechanics (QM/MM) analysis yielded an atomic description of the hydrolysis step for Mtb PPiase and, unexpectedly, indicated that Asp89, rather than Asp54 that was proposed for E. coli PPiase, can abstract a proton from a water molecule to activate it for a nucleophilic attack on the PPi substrate. Mutagenesis studies of the key Asp residues of Mtb PPiase supported this mechanism. This combination of structural and computational analyses clarifies our understanding of the mechanism of family I PPiases and has potential utility for rational development of drugs targeting this enzyme.
Categories: Journal Articles
Alignment of cryo-EM movies of individual particles by optimization of image translations
Publication date: Available online 19 August 2015
Source:Journal of Structural Biology
Author(s): John L. Rubinstein, Marcus A. Brubaker
Direct detector device (DDD) cameras have revolutionized single particle electron cryomicroscopy (cryo-EM). In addition to an improved camera detective quantum efficiency, acquisition of DDD movies allows for correction of movement of the specimen, due to both instabilities in the microscope specimen stage and electron beam-induced movement. Unlike specimen stage drift, beam-induced movement is not always homogeneous within an image. Local correlation in the trajectories of nearby particles suggests that beam-induced motion is due to deformation of the ice layer. Algorithms have already been described that can correct movement for large regions of frames and for >1MDa protein particles. Another algorithm allows individual <1MDa protein particle trajectories to be estimated, but requires rolling averages to be calculated from frames and fits linear trajectories for particles. Here we describe an algorithm that allows for individual <1MDa particle images to be aligned without frame averaging or linear trajectories. The algorithm maximizes the overall correlation of the shifted frames with the sum of the shifted frames. The optimum in this single objective function is found efficiently by making use of analytically calculated derivatives of the function. To smooth estimates of particle trajectories, rapid changes in particle positions between frames are penalized in the objective function and weighted averaging of nearby trajectories ensures local correlation in trajectories. This individual particle motion correction, in combination with weighting of Fourier components to account for increasing radiation damage in later frames, can be used to improve 3-D maps from single particle cryo-EM.
Source:Journal of Structural Biology
Author(s): John L. Rubinstein, Marcus A. Brubaker
Direct detector device (DDD) cameras have revolutionized single particle electron cryomicroscopy (cryo-EM). In addition to an improved camera detective quantum efficiency, acquisition of DDD movies allows for correction of movement of the specimen, due to both instabilities in the microscope specimen stage and electron beam-induced movement. Unlike specimen stage drift, beam-induced movement is not always homogeneous within an image. Local correlation in the trajectories of nearby particles suggests that beam-induced motion is due to deformation of the ice layer. Algorithms have already been described that can correct movement for large regions of frames and for >1MDa protein particles. Another algorithm allows individual <1MDa protein particle trajectories to be estimated, but requires rolling averages to be calculated from frames and fits linear trajectories for particles. Here we describe an algorithm that allows for individual <1MDa particle images to be aligned without frame averaging or linear trajectories. The algorithm maximizes the overall correlation of the shifted frames with the sum of the shifted frames. The optimum in this single objective function is found efficiently by making use of analytically calculated derivatives of the function. To smooth estimates of particle trajectories, rapid changes in particle positions between frames are penalized in the objective function and weighted averaging of nearby trajectories ensures local correlation in trajectories. This individual particle motion correction, in combination with weighting of Fourier components to account for increasing radiation damage in later frames, can be used to improve 3-D maps from single particle cryo-EM.
Categories: Journal Articles
Polyhedra structures and the evolution of the insect viruses
Publication date: Available online 18 August 2015
Source:Journal of Structural Biology
Author(s): Xiaoyun Ji, Danny Axford, Robin Owen, Gwyndaf Evans, Helen M. Ginn, Geoff Sutton, David I. Stuart
Polyhedra represent an ancient system used by a number of insect viruses to protect virions during long periods of environmental exposure. We present high resolution crystal structures of polyhedra for seven previously uncharacterised types of cypoviruses, four using ab initio selenomethionine phasing (two of these required over 100 selenomethionine crystals each). Approximately 80% of residues are structurally equivalent between all polyhedrins (pairwise rmsd ⩽1.5Å), whilst pairwise sequence identities, based on structural alignment, are as little as 12%. These structures illustrate the effect of 400million years of evolution on a system where the crystal lattice is the functionally conserved feature in the face of massive sequence variability. The conservation of crystal contacts is maintained across most of the molecular surface, except for a dispensable virus recognition domain. By spreading the contacts over so much of the protein surface the lattice remains robust in the face of many individual changes. Overall these unusual structural constraints seem to have skewed the molecule’s evolution so that surface residues are almost as conserved as the internal residues.
Source:Journal of Structural Biology
Author(s): Xiaoyun Ji, Danny Axford, Robin Owen, Gwyndaf Evans, Helen M. Ginn, Geoff Sutton, David I. Stuart
Polyhedra represent an ancient system used by a number of insect viruses to protect virions during long periods of environmental exposure. We present high resolution crystal structures of polyhedra for seven previously uncharacterised types of cypoviruses, four using ab initio selenomethionine phasing (two of these required over 100 selenomethionine crystals each). Approximately 80% of residues are structurally equivalent between all polyhedrins (pairwise rmsd ⩽1.5Å), whilst pairwise sequence identities, based on structural alignment, are as little as 12%. These structures illustrate the effect of 400million years of evolution on a system where the crystal lattice is the functionally conserved feature in the face of massive sequence variability. The conservation of crystal contacts is maintained across most of the molecular surface, except for a dispensable virus recognition domain. By spreading the contacts over so much of the protein surface the lattice remains robust in the face of many individual changes. Overall these unusual structural constraints seem to have skewed the molecule’s evolution so that surface residues are almost as conserved as the internal residues.
Categories: Journal Articles
Multi-scale simulation of plant stem reinforcement by brachysclereids: A case study in apple fruit peduncles
Publication date: Available online 13 August 2015
Source:Journal of Structural Biology
Author(s): Melanie Horbens, Dominik Branke, Roland Gärtner, Axel Voigt, Florian Stenger, Christoph Neinhuis
Sclereid formation in addition to or in gaps of fragmented fibre rings is common in dicotyledonous plant stems. Whether this sclereid formation is force-triggered remains open so far. In fruit peduncles of several Malus species as modified plant stems, for example, the persistent fibre ring is displaced to the centre by formation of cortex parenchyma during growth. Parenchyma cells subsequently differentiate into an additional layer of brachysclereids, previously interpreted as an adaptation to continuously rising fruit loads. The present study pursues a multi-scale numerical modelling approach, to verify the important effect for different cellular architectures in both sclerenchyma categories on the stiffness of these tissues and the entire peduncle. First, different material properties are simulated analogue to plant tissues on the basis of three cell types. A regular three-dimensional and a random Voronoi microstructure combined with various mechanical cell wall parameters are applied. Using homogenisation simulations based on HILL’s principle, numerical calculations predict a lower effective homogenised tissue stiffness of isodiametric brachysclereids compared to those of fibres, confirming experimentally obtained data from Malus fruit peduncles. Furthermore, a curved peduncle model with a complex arrangement of different material layers is generated. Diverse material sets are tested under three representative loadings, using an adaptive diffuse domain approach (AMDiS). The model explains the function of sclereids as considerable contributors to the stiffness against bending and tensile deformations, as well as torsion, especially in consequence of superimposed load conditions in the case of a curved plant stem.
Source:Journal of Structural Biology
Author(s): Melanie Horbens, Dominik Branke, Roland Gärtner, Axel Voigt, Florian Stenger, Christoph Neinhuis
Sclereid formation in addition to or in gaps of fragmented fibre rings is common in dicotyledonous plant stems. Whether this sclereid formation is force-triggered remains open so far. In fruit peduncles of several Malus species as modified plant stems, for example, the persistent fibre ring is displaced to the centre by formation of cortex parenchyma during growth. Parenchyma cells subsequently differentiate into an additional layer of brachysclereids, previously interpreted as an adaptation to continuously rising fruit loads. The present study pursues a multi-scale numerical modelling approach, to verify the important effect for different cellular architectures in both sclerenchyma categories on the stiffness of these tissues and the entire peduncle. First, different material properties are simulated analogue to plant tissues on the basis of three cell types. A regular three-dimensional and a random Voronoi microstructure combined with various mechanical cell wall parameters are applied. Using homogenisation simulations based on HILL’s principle, numerical calculations predict a lower effective homogenised tissue stiffness of isodiametric brachysclereids compared to those of fibres, confirming experimentally obtained data from Malus fruit peduncles. Furthermore, a curved peduncle model with a complex arrangement of different material layers is generated. Diverse material sets are tested under three representative loadings, using an adaptive diffuse domain approach (AMDiS). The model explains the function of sclereids as considerable contributors to the stiffness against bending and tensile deformations, as well as torsion, especially in consequence of superimposed load conditions in the case of a curved plant stem.
Categories: Journal Articles
Automatic estimation and correction of anisotropic magnification distortion in electron microscopes
Publication date: Available online 13 August 2015
Source:Journal of Structural Biology
Author(s): Timothy Grant, Nikolaus Grigorieff
We demonstrate a significant anisotropic magnification distortion, found on an FEI Titan Krios microscope and affecting magnifications commonly used for data acquisition on a Gatan K2 Summit detector. We describe a program (mag_distortion_estimate) to automatically estimate anisotropic magnification distortion from a set of images of a standard gold shadowed diffraction grating. We also describe a program (mag_distortion_correct) to correct for the estimated distortion in collected images. We demonstrate that the distortion present on the Titan Krios microscope limits the resolution of a set of rotavirus VP6 images to ∼7 Å, which increases to ∼3 Å following estimation and correction of the distortion. We also use a 70S ribosome sample to demonstrate that in addition to affecting resolution, magnification distortion can also interfere with the classification of heterogeneous data.
Source:Journal of Structural Biology
Author(s): Timothy Grant, Nikolaus Grigorieff
We demonstrate a significant anisotropic magnification distortion, found on an FEI Titan Krios microscope and affecting magnifications commonly used for data acquisition on a Gatan K2 Summit detector. We describe a program (mag_distortion_estimate) to automatically estimate anisotropic magnification distortion from a set of images of a standard gold shadowed diffraction grating. We also describe a program (mag_distortion_correct) to correct for the estimated distortion in collected images. We demonstrate that the distortion present on the Titan Krios microscope limits the resolution of a set of rotavirus VP6 images to ∼7 Å, which increases to ∼3 Å following estimation and correction of the distortion. We also use a 70S ribosome sample to demonstrate that in addition to affecting resolution, magnification distortion can also interfere with the classification of heterogeneous data.
Categories: Journal Articles
CTFFIND4: Fast and accurate defocus estimation from electron micrographs
Publication date: Available online 13 August 2015
Source:Journal of Structural Biology
Author(s): Alexis Rohou, Nikolaus Grigorieff
CTFFIND is a widely-used program for the estimation of objective lens defocus parameters from transmission electron micrographs. Defocus parameters are estimated by fitting a model of the microscope’s contrast transfer function (CTF) to an image’s amplitude spectrum. Here we describe modifications to the algorithm which make it significantly faster and more suitable for use with images collected using modern technologies such as dose fractionation and phase plates. We show that this new version preserves the accuracy of the original algorithm while allowing for higher throughput. We also describe a measure of the quality of the fit as a function of spatial frequency and suggest this can be used to define the highest resolution at which CTF oscillations were successfully modeled.
Source:Journal of Structural Biology
Author(s): Alexis Rohou, Nikolaus Grigorieff
CTFFIND is a widely-used program for the estimation of objective lens defocus parameters from transmission electron micrographs. Defocus parameters are estimated by fitting a model of the microscope’s contrast transfer function (CTF) to an image’s amplitude spectrum. Here we describe modifications to the algorithm which make it significantly faster and more suitable for use with images collected using modern technologies such as dose fractionation and phase plates. We show that this new version preserves the accuracy of the original algorithm while allowing for higher throughput. We also describe a measure of the quality of the fit as a function of spatial frequency and suggest this can be used to define the highest resolution at which CTF oscillations were successfully modeled.
Categories: Journal Articles
Structural and biochemical characterization of GTP cyclohydrolase II from Helicobacter pylori reveals its redox dependent catalytic activity
Publication date: Available online 10 August 2015
Source:Journal of Structural Biology
Author(s): Savita Yadav, Subramanian Karthikeyan
GTP cyclohydrolase II (GCHII), catalyzes the conversion of GTP to 2,5-diamino-6-β-ribosyl-4(3H)-pyrimidinone-5′-phosphate and has been shown to be essential for pathogens. Here we describe the biochemical, kinetic and structural characterization of GCHII from Helicobacter pylori (hGCHII). The crystal structure of hGCHII, unlike other GCHII structures, revealed that cysteines at the active site existed in oxidized state forming two disulfide bonds and lacked Zn2+ that was shown to be indispensable for catalytic activity in other species. However, incubation of hGCHII with hydrogen peroxide, an oxidizing agent, followed by PAR-assay showed that Zn2+ was intrinsically present, indicating that all cysteines at the catalytic site remained in reduced state. Moreover, site directed mutagenesis of catalytic site cysteines revealed that only three, out of four cysteines were essential for hGCHII activity. These results, though, indicated that hGCHII crystallized in oxidized form, the expulsion of Zn2+ upon oxidation of catalytic cysteines revealed its ability to act in response to the redox environment. Exploring further, incubation of hGCHII with reversible thiol modifying agent S-methyl-methane-thiosulfonate resulted in loss of GCHII activity due to oxidation of its cysteine residues as revealed by mass spectrometry studies. However, addition of reducing agent DTT partially restored the hGCHII catalytic activity. Taken together, these results demonstrate that hGCHII can regulate its catalytic activity depending on the redox environment, a function hitherto unknown for GCHII.
Source:Journal of Structural Biology
Author(s): Savita Yadav, Subramanian Karthikeyan
GTP cyclohydrolase II (GCHII), catalyzes the conversion of GTP to 2,5-diamino-6-β-ribosyl-4(3H)-pyrimidinone-5′-phosphate and has been shown to be essential for pathogens. Here we describe the biochemical, kinetic and structural characterization of GCHII from Helicobacter pylori (hGCHII). The crystal structure of hGCHII, unlike other GCHII structures, revealed that cysteines at the active site existed in oxidized state forming two disulfide bonds and lacked Zn2+ that was shown to be indispensable for catalytic activity in other species. However, incubation of hGCHII with hydrogen peroxide, an oxidizing agent, followed by PAR-assay showed that Zn2+ was intrinsically present, indicating that all cysteines at the catalytic site remained in reduced state. Moreover, site directed mutagenesis of catalytic site cysteines revealed that only three, out of four cysteines were essential for hGCHII activity. These results, though, indicated that hGCHII crystallized in oxidized form, the expulsion of Zn2+ upon oxidation of catalytic cysteines revealed its ability to act in response to the redox environment. Exploring further, incubation of hGCHII with reversible thiol modifying agent S-methyl-methane-thiosulfonate resulted in loss of GCHII activity due to oxidation of its cysteine residues as revealed by mass spectrometry studies. However, addition of reducing agent DTT partially restored the hGCHII catalytic activity. Taken together, these results demonstrate that hGCHII can regulate its catalytic activity depending on the redox environment, a function hitherto unknown for GCHII.
Categories: Journal Articles
Cover 2 - Editorial Board
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Source:Journal of Structural Biology, Volume 191, Issue 2
Categories: Journal Articles
Table of Contents / barcode
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Source:Journal of Structural Biology, Volume 191, Issue 2
Categories: Journal Articles
Structural and functional characterization of two unusual endonuclease III enzymes from Deinococcus radiodurans
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Aili Sarre, Mats Ökvist, Tobias Klar, David R. Hall, Arne O. Smalås, Sean McSweeney, Joanna Timmins, Elin Moe
While most bacteria possess a single gene encoding the bifunctional DNA glycosylase Endonuclease III (EndoIII) in their genomes, Deinococcus radiodurans possesses three: DR2438 (DrEndoIII1), DR0289 (DrEndoIII2) and DR0982 (DrEndoIII3). Here we have determined the crystal structures of DrEndoIII1 and an N-terminally truncated form of DrEndoIII3 (DrEndoIII3Δ76). We have also generated a homology model of DrEndoIII2 and measured activity of the three enzymes. All three structures consist of two all α-helical domains, one of which exhibits a [4Fe-4S] cluster and the other a HhH-motif, separated by a DNA binding cleft, similar to previously determined structures of endonuclease III from Escherichia coli and Geobacillus stearothermophilus. However, both DrEndoIII1 and DrEndoIII3 possess an extended HhH motif with extra helical features and an altered electrostatic surface potential. In addition, the DNA binding cleft of DrEndoIII3 seems to be less accessible for DNA interactions, while in DrEndoIII1 it seems to be more open. Analysis of the enzyme activities shows that DrEndoIII2 is most similar to the previously studied enzymes, while DrEndoIII1 seems to be more distant with a weaker activity towards substrate DNA containing either thymine glycol or an abasic site. DrEndoIII3 is the most distantly related enzyme and displays no detectable activity towards these substrates even though the suggested catalytic residues are conserved. Based on a comparative structural analysis, we suggest that the altered surface potential, shape of the substrate-binding pockets and specific amino acid substitutions close to the active site and in the DNA interacting loops may underlie the unexpected differences in activity.
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Aili Sarre, Mats Ökvist, Tobias Klar, David R. Hall, Arne O. Smalås, Sean McSweeney, Joanna Timmins, Elin Moe
While most bacteria possess a single gene encoding the bifunctional DNA glycosylase Endonuclease III (EndoIII) in their genomes, Deinococcus radiodurans possesses three: DR2438 (DrEndoIII1), DR0289 (DrEndoIII2) and DR0982 (DrEndoIII3). Here we have determined the crystal structures of DrEndoIII1 and an N-terminally truncated form of DrEndoIII3 (DrEndoIII3Δ76). We have also generated a homology model of DrEndoIII2 and measured activity of the three enzymes. All three structures consist of two all α-helical domains, one of which exhibits a [4Fe-4S] cluster and the other a HhH-motif, separated by a DNA binding cleft, similar to previously determined structures of endonuclease III from Escherichia coli and Geobacillus stearothermophilus. However, both DrEndoIII1 and DrEndoIII3 possess an extended HhH motif with extra helical features and an altered electrostatic surface potential. In addition, the DNA binding cleft of DrEndoIII3 seems to be less accessible for DNA interactions, while in DrEndoIII1 it seems to be more open. Analysis of the enzyme activities shows that DrEndoIII2 is most similar to the previously studied enzymes, while DrEndoIII1 seems to be more distant with a weaker activity towards substrate DNA containing either thymine glycol or an abasic site. DrEndoIII3 is the most distantly related enzyme and displays no detectable activity towards these substrates even though the suggested catalytic residues are conserved. Based on a comparative structural analysis, we suggest that the altered surface potential, shape of the substrate-binding pockets and specific amino acid substitutions close to the active site and in the DNA interacting loops may underlie the unexpected differences in activity.
Categories: Journal Articles
The N-terminal domain of MuB protein has striking structural similarity to DNA-binding domains and mediates MuB filament–filament interactions
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Marija Dramićanin, Blanca López-Méndez, Jasminka Boskovic, Ramón Campos-Olivas, Santiago Ramón-Maiques
MuB is an ATP-dependent DNA-binding protein that regulates the activity of MuA transposase and delivers the target DNA for transposition of phage Mu. Mechanistic insight into MuB function is limited to its AAA+ ATPase module, which upon ATP binding assembles into helical filaments around the DNA. However, the structure and function of the flexible N-terminal domain (NTD) appended to the AAA+ module remains uncharacterized. Here we report the solution structure of MuB NTD determined by NMR spectroscopy. The structure reveals a compact domain formed by four α-helices connected by short loops, and confirms the presence of a helix-turn-helix motif. High structural similarity and sequence homology with λ repressor-like DNA-binding domains suggest a possible role of MuB NTD in DNA binding. We also demonstrate that the NTD directly mediates the ability of MuB to establish filament–filament interactions. These findings lead us to a model in which the NTD interacts with the AAA+ spirals and perhaps also with the DNA bound within the filament, favoring MuB polymerization and filament clustering. We propose that the MuB NTD-dependent filament interactions might be an effective mechanism to bridge distant DNA regions during Mu transposition.
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Marija Dramićanin, Blanca López-Méndez, Jasminka Boskovic, Ramón Campos-Olivas, Santiago Ramón-Maiques
MuB is an ATP-dependent DNA-binding protein that regulates the activity of MuA transposase and delivers the target DNA for transposition of phage Mu. Mechanistic insight into MuB function is limited to its AAA+ ATPase module, which upon ATP binding assembles into helical filaments around the DNA. However, the structure and function of the flexible N-terminal domain (NTD) appended to the AAA+ module remains uncharacterized. Here we report the solution structure of MuB NTD determined by NMR spectroscopy. The structure reveals a compact domain formed by four α-helices connected by short loops, and confirms the presence of a helix-turn-helix motif. High structural similarity and sequence homology with λ repressor-like DNA-binding domains suggest a possible role of MuB NTD in DNA binding. We also demonstrate that the NTD directly mediates the ability of MuB to establish filament–filament interactions. These findings lead us to a model in which the NTD interacts with the AAA+ spirals and perhaps also with the DNA bound within the filament, favoring MuB polymerization and filament clustering. We propose that the MuB NTD-dependent filament interactions might be an effective mechanism to bridge distant DNA regions during Mu transposition.
Categories: Journal Articles
Computer-aided design of aptamers for cytochrome p450
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Dmitrii S. Shcherbinin, Oksana V. Gnedenko, Svetlana A. Khmeleva, Sergey A. Usanov, Andrei A. Gilep, Aliaksei V. Yantsevich, Tatsiana V. Shkel, Ivan V. Yushkevich, Sergey P. Radko, Alexis S. Ivanov, Alexander V. Veselovsky, Alexander I. Archakov
Aptamers are short single-stranded DNA or RNA oligonucleotides that can bind to their targets with high affinity and specificity. Usually, they are experimentally selected using the SELEX method. Here, we describe an approach toward the in silico selection of aptamers for proteins. This approach involves three steps: finding a potential binding site, designing the recognition and structural parts of the aptamers and evaluating the experimental affinity. Using this approach, a set of 15-mer aptamers for cytochrome P450 51A1 was designed using docking and molecular dynamics simulation. An experimental evaluation of the synthesized aptamers using SPR biosensor showed that these aptamers interact with cytochrome P450 51A1 with K d values in the range of 10−6–10−7 M.
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Dmitrii S. Shcherbinin, Oksana V. Gnedenko, Svetlana A. Khmeleva, Sergey A. Usanov, Andrei A. Gilep, Aliaksei V. Yantsevich, Tatsiana V. Shkel, Ivan V. Yushkevich, Sergey P. Radko, Alexis S. Ivanov, Alexander V. Veselovsky, Alexander I. Archakov
Aptamers are short single-stranded DNA or RNA oligonucleotides that can bind to their targets with high affinity and specificity. Usually, they are experimentally selected using the SELEX method. Here, we describe an approach toward the in silico selection of aptamers for proteins. This approach involves three steps: finding a potential binding site, designing the recognition and structural parts of the aptamers and evaluating the experimental affinity. Using this approach, a set of 15-mer aptamers for cytochrome P450 51A1 was designed using docking and molecular dynamics simulation. An experimental evaluation of the synthesized aptamers using SPR biosensor showed that these aptamers interact with cytochrome P450 51A1 with K d values in the range of 10−6–10−7 M.
Categories: Journal Articles
Molecular dynamics simulation study reveals potential substrate entry path into γ-secretase/presenilin-1
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Ren Kong, Shan Chang, Weiming Xia, Stephen T.C. Wong
Presenilin 1 (PS1) is the catalytic unit of γ-secretase which cleaves more than one hundred substrates. Among them, amyloid precursor protein (APP) and Notch are notable for their pivotal role in the pathogenesis of Alzheimer’s disease (AD) and certain types of cancer. The hydrolysis process occurring inside the hydrophobic lipid bilayer remains unclear. With the aim to understand the mechanism of intramembrane proteolysis by γ-secretase, we constructed a homology model of human PS1 and performed molecular dynamics simulation in explicit membrane phospholipids with different components. During the simulation, TM9 was found to exhibit a high level of flexibility that involved in “gate-open” movement of TM2 and TM6, and thus partially exposed the catalytic residues. The highly conserved PALP motif acts as an anchor to mediate the conformation changes of TM6 induced by TM9. Moreover, direct interactions were observed between 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and the active site of γ-secretase, indicating that the lipid molecules have the potential to modulate γ-secretase by contacting with the catalytic residues, i.e., ASP 257 and ASP 385 of PS1. The intermediate states indicate a potential substrate penetration pathway through the interface of TM2 and TM6, which may be induced by changes of TM9. To our knowledge, this is the first molecular simulation study that reveals dynamic behavior of the human PS1 structure in the lipid bilayer and provides insight into the substrate entry path for subsequent intramembrane hydrolysis, which is critical information required for new strategy development of γ-secretase modulators to alleviate devastating AD.
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Ren Kong, Shan Chang, Weiming Xia, Stephen T.C. Wong
Presenilin 1 (PS1) is the catalytic unit of γ-secretase which cleaves more than one hundred substrates. Among them, amyloid precursor protein (APP) and Notch are notable for their pivotal role in the pathogenesis of Alzheimer’s disease (AD) and certain types of cancer. The hydrolysis process occurring inside the hydrophobic lipid bilayer remains unclear. With the aim to understand the mechanism of intramembrane proteolysis by γ-secretase, we constructed a homology model of human PS1 and performed molecular dynamics simulation in explicit membrane phospholipids with different components. During the simulation, TM9 was found to exhibit a high level of flexibility that involved in “gate-open” movement of TM2 and TM6, and thus partially exposed the catalytic residues. The highly conserved PALP motif acts as an anchor to mediate the conformation changes of TM6 induced by TM9. Moreover, direct interactions were observed between 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and the active site of γ-secretase, indicating that the lipid molecules have the potential to modulate γ-secretase by contacting with the catalytic residues, i.e., ASP 257 and ASP 385 of PS1. The intermediate states indicate a potential substrate penetration pathway through the interface of TM2 and TM6, which may be induced by changes of TM9. To our knowledge, this is the first molecular simulation study that reveals dynamic behavior of the human PS1 structure in the lipid bilayer and provides insight into the substrate entry path for subsequent intramembrane hydrolysis, which is critical information required for new strategy development of γ-secretase modulators to alleviate devastating AD.
Categories: Journal Articles
Ion and seed dependent fibril assembly of a spidroin core domain
Publication date: August 2015
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Martin Humenik, Andrew M. Smith, Sina Arndt, Thomas Scheibel
Recombinant eADF4(C16) represents an engineered spider silk variant based on the sequence of the core domain of the natural dragline silk protein ADF4 of Araneus diadematus. Previously eADF4(C16) has been shown to self-assemble into cross-β fibrils in a two-step process of nucleus formation and fibril growth. Here, it is shown that structurally converted low molecular weight oligomers can act as nuclei. Further, it could be determined that specifically potassium and phosphate ions strongly influence both nucleus formation as well as fibril growth. Nucleation of fibril assembly could be surpassed by seeding soluble protein with pre-assembled fibrils but also, unexpectedly, with eADF4(C16) sub-micrometer particles. The latter finding reveals that spider silk fibril assembly seems to be rather dependent on the protein sequence than on the structural features, since cross-seeding with other proteins was not possible.
Source:Journal of Structural Biology, Volume 191, Issue 2
Author(s): Martin Humenik, Andrew M. Smith, Sina Arndt, Thomas Scheibel
Recombinant eADF4(C16) represents an engineered spider silk variant based on the sequence of the core domain of the natural dragline silk protein ADF4 of Araneus diadematus. Previously eADF4(C16) has been shown to self-assemble into cross-β fibrils in a two-step process of nucleus formation and fibril growth. Here, it is shown that structurally converted low molecular weight oligomers can act as nuclei. Further, it could be determined that specifically potassium and phosphate ions strongly influence both nucleus formation as well as fibril growth. Nucleation of fibril assembly could be surpassed by seeding soluble protein with pre-assembled fibrils but also, unexpectedly, with eADF4(C16) sub-micrometer particles. The latter finding reveals that spider silk fibril assembly seems to be rather dependent on the protein sequence than on the structural features, since cross-seeding with other proteins was not possible.
Categories: Journal Articles