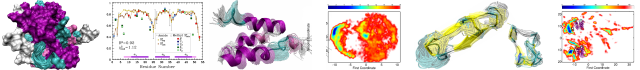

Journal Articles
Quantitative selection analysis of bacteriophage φCbK susceptibility in Caulobacter crescentus
Publication date: Available online 22 November 2015
Source:Journal of Molecular Biology
Author(s): Matthias Christen, Christian Beusch, Yvonne Bösch, Dario Cerletti, Carlos Eduardo Flores-Tinoco, Luca Del Medico, Flavia Tschan, Beat Christen
Classical molecular genetics uses stringent selective conditions to identify mutants with distinct phenotypic responses. Mutations giving rise to less pronounced phenotypes are often missed. However, to gain systems-level insights into complex genetic interaction networks requires genome-wide assignment of quantitative phenotypic traits. In this paper, we present a quantitative selection approach coupled with transposon sequencing (QS-TnSeq) to globally identify the cellular components that orchestrate susceptibility of the cell-cycle model bacterium Caulobacter crescentus towards bacteriophage φCbK infection. We found that 135 genes representing 3.30% of the Caulobacter genome exhibit significant accumulation of transposon insertions upon φCbK selection. More than 85% thereof consist of new factors not previously associated with phage φCbK susceptibility. Using hierarchical clustering of dose-dependent TnSeq datasets, we grouped these genes into functional modules that correlate with different stages of the φCbK infection process. We assign φCbK susceptibility to eight new genes that represent novel components of the pilus secretion machinery. Further, we demonstrate that from 86 motility genes, only seven genes encoding structural and regulatory components of the flagellar hook increase phage resistance when disrupted by transposons, suggesting a link between flagellar hook assembly and pili biogenesis. In addition, we observe high recovery of Tn5 insertions within regulatory sequences of the genes encoding the essential NADH:ubiquinone oxidoreductase complex indicating that intact proton motive force is crucial for effective phage propagation. In sum, QS-TnSeq is broadly applicable to perform quantitative and genome-wide systems-genetics analysis of complex phenotypic traits.
Graphical abstract
Source:Journal of Molecular Biology
Author(s): Matthias Christen, Christian Beusch, Yvonne Bösch, Dario Cerletti, Carlos Eduardo Flores-Tinoco, Luca Del Medico, Flavia Tschan, Beat Christen
Classical molecular genetics uses stringent selective conditions to identify mutants with distinct phenotypic responses. Mutations giving rise to less pronounced phenotypes are often missed. However, to gain systems-level insights into complex genetic interaction networks requires genome-wide assignment of quantitative phenotypic traits. In this paper, we present a quantitative selection approach coupled with transposon sequencing (QS-TnSeq) to globally identify the cellular components that orchestrate susceptibility of the cell-cycle model bacterium Caulobacter crescentus towards bacteriophage φCbK infection. We found that 135 genes representing 3.30% of the Caulobacter genome exhibit significant accumulation of transposon insertions upon φCbK selection. More than 85% thereof consist of new factors not previously associated with phage φCbK susceptibility. Using hierarchical clustering of dose-dependent TnSeq datasets, we grouped these genes into functional modules that correlate with different stages of the φCbK infection process. We assign φCbK susceptibility to eight new genes that represent novel components of the pilus secretion machinery. Further, we demonstrate that from 86 motility genes, only seven genes encoding structural and regulatory components of the flagellar hook increase phage resistance when disrupted by transposons, suggesting a link between flagellar hook assembly and pili biogenesis. In addition, we observe high recovery of Tn5 insertions within regulatory sequences of the genes encoding the essential NADH:ubiquinone oxidoreductase complex indicating that intact proton motive force is crucial for effective phage propagation. In sum, QS-TnSeq is broadly applicable to perform quantitative and genome-wide systems-genetics analysis of complex phenotypic traits.
Graphical abstract
Categories: Journal Articles
Editorial Board
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Source:Journal of Molecular Biology, Volume 427, Issue 23
Categories: Journal Articles
Contents List
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Source:Journal of Molecular Biology, Volume 427, Issue 23
Categories: Journal Articles
A Snapshot of the Extraordinary World of Social Microbiology
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Nicola R. Stanley-Wall, Sarah J. Coulthurst, Ian Barry Holland
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Nicola R. Stanley-Wall, Sarah J. Coulthurst, Ian Barry Holland
Categories: Journal Articles
Pseudomonas aeruginosa Biofilm Infections: Community Structure, Antimicrobial Tolerance and Immune Response
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Morten Rybtke, Louise Dahl Hultqvist, Michael Givskov, Tim Tolker-Nielsen
Studies of biopsies from infectious sites, explanted tissue and medical devises have provided evidence that biofilms are the underlying cause of a variety of tissue-associated and implant-associated recalcitrant human infections. With a need for novel anti-biofilm treatment strategies, research in biofilm infection microbiology, biofilm formation mechanisms and biofilm-associated antimicrobial tolerance has become an important area in microbiology. Substantial knowledge about biofilm formation mechanisms, biofilm-associated antimicrobial tolerance and immune evasion mechanisms has been obtained through work with biofilms grown in in vitro experimental setups, and the relevance of this information in the context of chronic infections is being investigated by the use of animal models of infection. Because our current in vitro experimental setups and animal models have limitations, new advanced in vitro models developed with knowledge about the chemical landscape at infectious sites are needed.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Morten Rybtke, Louise Dahl Hultqvist, Michael Givskov, Tim Tolker-Nielsen
Studies of biopsies from infectious sites, explanted tissue and medical devises have provided evidence that biofilms are the underlying cause of a variety of tissue-associated and implant-associated recalcitrant human infections. With a need for novel anti-biofilm treatment strategies, research in biofilm infection microbiology, biofilm formation mechanisms and biofilm-associated antimicrobial tolerance has become an important area in microbiology. Substantial knowledge about biofilm formation mechanisms, biofilm-associated antimicrobial tolerance and immune evasion mechanisms has been obtained through work with biofilms grown in in vitro experimental setups, and the relevance of this information in the context of chronic infections is being investigated by the use of animal models of infection. Because our current in vitro experimental setups and animal models have limitations, new advanced in vitro models developed with knowledge about the chemical landscape at infectious sites are needed.
Graphical abstract
Categories: Journal Articles
The Limitations of In Vitro Experimentation in Understanding Biofilms and Chronic Infection
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Aled E.L. Roberts, Kasper N. Kragh, Thomas Bjarnsholt, Stephen P. Diggle
We have become increasingly aware that, during infection, pathogenic bacteria often grow in multicellular biofilms that are often highly resistant to antibacterial strategies. In order to understand how biofilms form and contribute to infection, many research groups around the world have heavily used in vitro biofilm systems such as microtitre plate assays and flow cells. Whilst these methods have greatly increased our understanding of the biology of biofilms, it is becoming increasingly apparent that many of our in vitro methods do not accurately represent in vivo conditions. Here we present a systematic review of the most widely used in vitro biofilm systems, and we discuss why they are not always representative of the in vivo biofilms found in chronic infections. We present examples of methods that will help us to bridge the gap between in vitro and in vivo biofilm work so that we can ultimately use our benchside data to improve bedside treatment.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Aled E.L. Roberts, Kasper N. Kragh, Thomas Bjarnsholt, Stephen P. Diggle
We have become increasingly aware that, during infection, pathogenic bacteria often grow in multicellular biofilms that are often highly resistant to antibacterial strategies. In order to understand how biofilms form and contribute to infection, many research groups around the world have heavily used in vitro biofilm systems such as microtitre plate assays and flow cells. Whilst these methods have greatly increased our understanding of the biology of biofilms, it is becoming increasingly apparent that many of our in vitro methods do not accurately represent in vivo conditions. Here we present a systematic review of the most widely used in vitro biofilm systems, and we discuss why they are not always representative of the in vivo biofilms found in chronic infections. We present examples of methods that will help us to bridge the gap between in vitro and in vivo biofilm work so that we can ultimately use our benchside data to improve bedside treatment.
Graphical abstract
Categories: Journal Articles
Intermicrobial Interactions as a Driver for Community Composition and Stratification of Oral Biofilms
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Nicholas S. Jakubovics
The oral cavity is accessible to microorganisms, and biofilms are present throughout on hard and soft tissues. The shedding of epithelial cell layers is usually effective for controlling biofilm development on soft tissues. Innate immune mechanisms are not so effective against biofilms on tooth surfaces, and oral hygiene measures such as brushing and flossing are required for the periodic removal of dental plaque. Even with good oral hygiene, microbial communities accumulate on teeth in areas that are protected from mechanical abrasion forces. Changes in the composition of these biofilms are associated with oral diseases such as dental caries or periodontitis. Newly formed biofilms and more mature dental plaque each have a level of spatial organization in the horizontal and vertical planes. Communities are shaped by many varied interactions between different species and genera within the biofilm, which include physical cell–cell associations known as coaggregation, interspecies signaling, secretion and turnover of antimicrobial compounds and the sharing of an extracellular matrix. Central to these interactions is the selection for metabolic synergies and it is becoming clear that the ability of communities to extract the maximum energy from the available metabolites is a potent driver for biofilm structure and stratification. This review discusses recent advances in our understanding of intermicrobial interactions in oral biofilms and the roles that they play in determining the spatial organization of biofilm communities.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Nicholas S. Jakubovics
The oral cavity is accessible to microorganisms, and biofilms are present throughout on hard and soft tissues. The shedding of epithelial cell layers is usually effective for controlling biofilm development on soft tissues. Innate immune mechanisms are not so effective against biofilms on tooth surfaces, and oral hygiene measures such as brushing and flossing are required for the periodic removal of dental plaque. Even with good oral hygiene, microbial communities accumulate on teeth in areas that are protected from mechanical abrasion forces. Changes in the composition of these biofilms are associated with oral diseases such as dental caries or periodontitis. Newly formed biofilms and more mature dental plaque each have a level of spatial organization in the horizontal and vertical planes. Communities are shaped by many varied interactions between different species and genera within the biofilm, which include physical cell–cell associations known as coaggregation, interspecies signaling, secretion and turnover of antimicrobial compounds and the sharing of an extracellular matrix. Central to these interactions is the selection for metabolic synergies and it is becoming clear that the ability of communities to extract the maximum energy from the available metabolites is a potent driver for biofilm structure and stratification. This review discusses recent advances in our understanding of intermicrobial interactions in oral biofilms and the roles that they play in determining the spatial organization of biofilm communities.
Graphical abstract
Categories: Journal Articles
Pathogen Resistance Mediated by IL-22 Signaling at the Epithelial–Microbiota Interface
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Fernanda Schreiber, Julia Maryam Arasteh, Trevor D. Lawley
Intestinal colonization resistance to bacterial pathogens is generally associated, among other factors, with mucosal homeostasis that preserves the integrity of the intestinal barrier. Mucosal homeostasis depends on physical and molecular interactions between three components: the resident microbiota, the epithelial layer and the local immune system. The cytokine IL-22 helps to orchestrate this three-way interaction. IL-22 is produced by immune cells present beneath the epithelium and is induced by bacteria present in the intestine. IL-22 stimulates the epithelial cells via the IL-22RA1–IL-10R2 receptor complex inducing changes in the expression of genes involved in the maintenance of epithelial barrier integrity, with a variety of functions in pathogen resistance such as mucus layer modifications and hydration, tight junction fortification and the production of a broad range of bactericidal compounds. These mechanisms of pathogen resistance, in turn, affect the microbiota composition and create an environment that excludes pathogens. Here we highlight the role of IL-22 as key mediator in the give-and-take relationship between the microbiota and the host that impacts pathogen resistance.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Fernanda Schreiber, Julia Maryam Arasteh, Trevor D. Lawley
Intestinal colonization resistance to bacterial pathogens is generally associated, among other factors, with mucosal homeostasis that preserves the integrity of the intestinal barrier. Mucosal homeostasis depends on physical and molecular interactions between three components: the resident microbiota, the epithelial layer and the local immune system. The cytokine IL-22 helps to orchestrate this three-way interaction. IL-22 is produced by immune cells present beneath the epithelium and is induced by bacteria present in the intestine. IL-22 stimulates the epithelial cells via the IL-22RA1–IL-10R2 receptor complex inducing changes in the expression of genes involved in the maintenance of epithelial barrier integrity, with a variety of functions in pathogen resistance such as mucus layer modifications and hydration, tight junction fortification and the production of a broad range of bactericidal compounds. These mechanisms of pathogen resistance, in turn, affect the microbiota composition and create an environment that excludes pathogens. Here we highlight the role of IL-22 as key mediator in the give-and-take relationship between the microbiota and the host that impacts pathogen resistance.
Graphical abstract
Categories: Journal Articles
Shelter in a Swarm
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Rasika M. Harshey, Jonathan D. Partridge
Flagella propel bacteria during both swimming and swarming, dispersing them widely. However, while swimming bacteria use chemotaxis to find nutrients and avoid toxic environments, swarming bacteria appear to suppress chemotaxis and to use the dynamics of their collective motion to continuously expand and acquire new territory, barrel through lethal chemicals in their path, carry along bacterial and fungal cargo that assists in exploration of new niches, and engage in group warfare for niche dominance. Here, we focus on two aspects of swarming, which, if understood, hold the promise of revealing new insights into microbial signaling and behavior, with ramifications beyond bacterial swarming. These are as follows: how bacteria sense they are on a surface and turn on programs that promote movement and how they override scarcity and adversity as dense packs.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Rasika M. Harshey, Jonathan D. Partridge
Flagella propel bacteria during both swimming and swarming, dispersing them widely. However, while swimming bacteria use chemotaxis to find nutrients and avoid toxic environments, swarming bacteria appear to suppress chemotaxis and to use the dynamics of their collective motion to continuously expand and acquire new territory, barrel through lethal chemicals in their path, carry along bacterial and fungal cargo that assists in exploration of new niches, and engage in group warfare for niche dominance. Here, we focus on two aspects of swarming, which, if understood, hold the promise of revealing new insights into microbial signaling and behavior, with ramifications beyond bacterial swarming. These are as follows: how bacteria sense they are on a surface and turn on programs that promote movement and how they override scarcity and adversity as dense packs.
Graphical abstract
Categories: Journal Articles
Motility, Chemotaxis and Aerotaxis Contribute to Competitiveness during Bacterial Pellicle Biofilm Development
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Theresa Hölscher, Benjamin Bartels, Yu-Cheng Lin, Ramses Gallegos-Monterrosa, Alexa Price-Whelan, Roberto Kolter, Lars E.P. Dietrich, Ákos T. Kovács
Biofilm formation is a complex process involving various signaling pathways and changes in gene expression. Many of the sensory mechanisms and regulatory cascades involved have been defined for biofilms formed by diverse organisms attached to solid surfaces. By comparison, our knowledge on the basic mechanisms underlying the formation of biofilms at air–liquid interfaces, that is, pellicles, is much less complete. In particular, the roles of flagella have been studied in multiple solid-surface biofilm models but remain largely undefined for pellicles. In this work, we characterize the contributions of flagellum-based motility, chemotaxis and oxygen sensing to pellicle formation in the Gram-positive Bacillus subtilis. We confirm that flagellum-based motility is involved in, but is not absolutely essential for, B. subtilis pellicle formation. Further, we show that flagellum-based motility, chemotaxis and oxygen sensing are important for successful competition during B. subtilis pellicle formation. We report that flagellum-based motility similarly contributes to pellicle formation and fitness in competition assays in the Gram-negative Pseudomonas aeruginosa. Time-lapse imaging of static liquid cultures demonstrates that, in both B. subtilis and P. aeruginosa, a turbulent flow forms in the tube and a zone of clearing appears below the air–liquid interface just before the formation of the pellicle but only in strains that have flagella.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Theresa Hölscher, Benjamin Bartels, Yu-Cheng Lin, Ramses Gallegos-Monterrosa, Alexa Price-Whelan, Roberto Kolter, Lars E.P. Dietrich, Ákos T. Kovács
Biofilm formation is a complex process involving various signaling pathways and changes in gene expression. Many of the sensory mechanisms and regulatory cascades involved have been defined for biofilms formed by diverse organisms attached to solid surfaces. By comparison, our knowledge on the basic mechanisms underlying the formation of biofilms at air–liquid interfaces, that is, pellicles, is much less complete. In particular, the roles of flagella have been studied in multiple solid-surface biofilm models but remain largely undefined for pellicles. In this work, we characterize the contributions of flagellum-based motility, chemotaxis and oxygen sensing to pellicle formation in the Gram-positive Bacillus subtilis. We confirm that flagellum-based motility is involved in, but is not absolutely essential for, B. subtilis pellicle formation. Further, we show that flagellum-based motility, chemotaxis and oxygen sensing are important for successful competition during B. subtilis pellicle formation. We report that flagellum-based motility similarly contributes to pellicle formation and fitness in competition assays in the Gram-negative Pseudomonas aeruginosa. Time-lapse imaging of static liquid cultures demonstrates that, in both B. subtilis and P. aeruginosa, a turbulent flow forms in the tube and a zone of clearing appears below the air–liquid interface just before the formation of the pellicle but only in strains that have flagella.
Graphical abstract
Categories: Journal Articles
How Myxobacteria Cooperate
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Pengbo Cao, Arup Dey, Christopher N. Vassallo, Daniel Wall
Prokaryotes often reside in groups where a high degree of relatedness has allowed the evolution of cooperative behaviors. However, very few bacteria or archaea have made the successful transition from unicellular to obligate multicellular life. A notable exception is the myxobacteria, in which cells cooperate to perform group functions highlighted by fruiting body development, an obligate multicellular function. Like all multicellular organisms, myxobacteria face challenges in how to organize and maintain multicellularity. These challenges include maintaining population homeostasis, carrying out tissue repair and regulating the behavior of non-cooperators. Here, we describe the major cooperative behaviors that myxobacteria use: motility, predation and development. In addition, this review emphasizes recent discoveries in the social behavior of outer membrane exchange, wherein kin share outer membrane contents. Finally, we review evidence that outer membrane exchange may be involved in regulating population homeostasis, thus serving as a social tool for myxobacteria to make the cyclic transitions from unicellular to multicellular states.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Pengbo Cao, Arup Dey, Christopher N. Vassallo, Daniel Wall
Prokaryotes often reside in groups where a high degree of relatedness has allowed the evolution of cooperative behaviors. However, very few bacteria or archaea have made the successful transition from unicellular to obligate multicellular life. A notable exception is the myxobacteria, in which cells cooperate to perform group functions highlighted by fruiting body development, an obligate multicellular function. Like all multicellular organisms, myxobacteria face challenges in how to organize and maintain multicellularity. These challenges include maintaining population homeostasis, carrying out tissue repair and regulating the behavior of non-cooperators. Here, we describe the major cooperative behaviors that myxobacteria use: motility, predation and development. In addition, this review emphasizes recent discoveries in the social behavior of outer membrane exchange, wherein kin share outer membrane contents. Finally, we review evidence that outer membrane exchange may be involved in regulating population homeostasis, thus serving as a social tool for myxobacteria to make the cyclic transitions from unicellular to multicellular states.
Graphical abstract
Categories: Journal Articles
The Evolution of Aggregative Multicellularity and Cell–Cell Communication in the Dictyostelia
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Qingyou Du, Yoshinori Kawabe, Christina Schilde, Zhi-hui Chen, Pauline Schaap
Aggregative multicellularity, resulting in formation of a spore-bearing fruiting body, evolved at least six times independently amongst both eukaryotes and prokaryotes. Amongst eukaryotes, this form of multicellularity is mainly studied in the social amoeba Dictyostelium discoideum. In this review, we summarise trends in the evolution of cell-type specialisation and behavioural complexity in the four major groups of Dictyostelia. We describe the cell–cell communication systems that control the developmental programme of D. discoideum, highlighting the central role of cAMP in the regulation of cell movement and cell differentiation. Comparative genomic studies showed that the proteins involved in cAMP signalling are deeply conserved across Dictyostelia and their unicellular amoebozoan ancestors. Comparative functional analysis revealed that cAMP signalling in D. discoideum originated from a second messenger role in amoebozoan encystation. We highlight some molecular changes in cAMP signalling genes that were responsible for the novel roles of cAMP in multicellular development.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Qingyou Du, Yoshinori Kawabe, Christina Schilde, Zhi-hui Chen, Pauline Schaap
Aggregative multicellularity, resulting in formation of a spore-bearing fruiting body, evolved at least six times independently amongst both eukaryotes and prokaryotes. Amongst eukaryotes, this form of multicellularity is mainly studied in the social amoeba Dictyostelium discoideum. In this review, we summarise trends in the evolution of cell-type specialisation and behavioural complexity in the four major groups of Dictyostelia. We describe the cell–cell communication systems that control the developmental programme of D. discoideum, highlighting the central role of cAMP in the regulation of cell movement and cell differentiation. Comparative genomic studies showed that the proteins involved in cAMP signalling are deeply conserved across Dictyostelia and their unicellular amoebozoan ancestors. Comparative functional analysis revealed that cAMP signalling in D. discoideum originated from a second messenger role in amoebozoan encystation. We highlight some molecular changes in cAMP signalling genes that were responsible for the novel roles of cAMP in multicellular development.
Graphical abstract
Categories: Journal Articles
Brainless but Multi-Headed: Decision Making by the Acellular Slime Mould Physarum polycephalum
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Madeleine Beekman, Tanya Latty
Because of its peculiar biology and the ease with which it can be cultured, the acellular slime mould Physarum polycephalum has long been a model organism in a range of disciplines. Due to its macroscopic, syncytial nature, it is no surprise that it has been a favourite amongst cell biologists. Its inclusion in the experimental tool kit of behavioural ecologists is much more recent. These recent studies have certainly paid off. They have shown that, for an organism that lacks a brain or central nervous system, P. polycephalum shows rather complex behaviour. For example, it is capable of finding the shortest path through a maze, it can construct networks as efficient as those designed by humans, it can solve computationally difficult puzzles, it makes multi-objective foraging decisions, it balances its nutrient intake and it even behaves irrationally. Are the slime mould's achievements simply “cute”, worthy of mentioning in passing but nothing to take too seriously? Or do they hint at the fundamental processes underlying all decision making? We will address this question after reviewing the decision-making abilities of the slime mould.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Madeleine Beekman, Tanya Latty
Because of its peculiar biology and the ease with which it can be cultured, the acellular slime mould Physarum polycephalum has long been a model organism in a range of disciplines. Due to its macroscopic, syncytial nature, it is no surprise that it has been a favourite amongst cell biologists. Its inclusion in the experimental tool kit of behavioural ecologists is much more recent. These recent studies have certainly paid off. They have shown that, for an organism that lacks a brain or central nervous system, P. polycephalum shows rather complex behaviour. For example, it is capable of finding the shortest path through a maze, it can construct networks as efficient as those designed by humans, it can solve computationally difficult puzzles, it makes multi-objective foraging decisions, it balances its nutrient intake and it even behaves irrationally. Are the slime mould's achievements simply “cute”, worthy of mentioning in passing but nothing to take too seriously? Or do they hint at the fundamental processes underlying all decision making? We will address this question after reviewing the decision-making abilities of the slime mould.
Graphical abstract
Categories: Journal Articles
Bacterial danger sensing
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Michele LeRoux, S. Brook Peterson, Joseph D. Mougous
Here we propose that bacteria detect and respond to threats posed by other bacteria via an innate immune-like process that we term danger sensing. We find support for this contention by reexamining existing literature from the perspective that intermicrobial antagonism, not opportunistic pathogenesis, is the major evolutionary force shaping the defensive behaviors of most bacteria. We conclude that many bacteria possess danger sensing pathways composed of a danger signal receptor and corresponding signal transduction mechanism that regulate pathways important for survival in the presence of the perceived competitor.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Michele LeRoux, S. Brook Peterson, Joseph D. Mougous
Here we propose that bacteria detect and respond to threats posed by other bacteria via an innate immune-like process that we term danger sensing. We find support for this contention by reexamining existing literature from the perspective that intermicrobial antagonism, not opportunistic pathogenesis, is the major evolutionary force shaping the defensive behaviors of most bacteria. We conclude that many bacteria possess danger sensing pathways composed of a danger signal receptor and corresponding signal transduction mechanism that regulate pathways important for survival in the presence of the perceived competitor.
Graphical abstract
Categories: Journal Articles
Contact-Dependent Growth Inhibition (CDI) and CdiB/CdiA Two-Partner Secretion Proteins
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Julia L.E. Willett, Zachary C. Ruhe, Celia W. Goulding, David A. Low, Christopher S. Hayes
Bacteria have developed several strategies to communicate and compete with one another in complex environments. One important mechanism of inter-bacterial competition is contact-dependent growth inhibition (CDI), in which Gram-negative bacteria use CdiB/CdiA two-partner secretion proteins to suppress the growth of neighboring target cells. CdiB is an Omp85 outer-membrane protein that exports and assembles CdiA exoproteins onto the inhibitor cell surface. CdiA binds to receptors on susceptible bacteria and subsequently delivers its C-terminal toxin domain (CdiA-CT) into the target cell. CDI systems also encode CdiI immunity proteins, which specifically bind to the CdiA-CT and neutralize its toxin activity, thereby protecting CDI+ cells from auto-inhibition. Remarkably, CdiA-CT sequences are highly variable between bacteria, as are the corresponding CdiI immunity proteins. Variations in CDI toxin/immunity proteins suggest that these systems function in bacterial self/non-self recognition and thereby play an important role in microbial communities. In this review, we discuss recent advances in the biochemistry, structural biology and physiology of CDI.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Julia L.E. Willett, Zachary C. Ruhe, Celia W. Goulding, David A. Low, Christopher S. Hayes
Bacteria have developed several strategies to communicate and compete with one another in complex environments. One important mechanism of inter-bacterial competition is contact-dependent growth inhibition (CDI), in which Gram-negative bacteria use CdiB/CdiA two-partner secretion proteins to suppress the growth of neighboring target cells. CdiB is an Omp85 outer-membrane protein that exports and assembles CdiA exoproteins onto the inhibitor cell surface. CdiA binds to receptors on susceptible bacteria and subsequently delivers its C-terminal toxin domain (CdiA-CT) into the target cell. CDI systems also encode CdiI immunity proteins, which specifically bind to the CdiA-CT and neutralize its toxin activity, thereby protecting CDI+ cells from auto-inhibition. Remarkably, CdiA-CT sequences are highly variable between bacteria, as are the corresponding CdiI immunity proteins. Variations in CDI toxin/immunity proteins suggest that these systems function in bacterial self/non-self recognition and thereby play an important role in microbial communities. In this review, we discuss recent advances in the biochemistry, structural biology and physiology of CDI.
Graphical abstract
Categories: Journal Articles
Diversification of β-Augmentation Interactions between CDI Toxin/Immunity Proteins
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Robert P. Morse, Julia L.E. Willett, Parker M. Johnson, Jing Zheng, Alfredo Credali, Angelina Iniguez, James S. Nowick, Christopher S. Hayes, Celia W. Goulding
Contact-dependent growth inhibition (CDI) is a widespread mechanism of inter-bacterial competition mediated by the CdiB/CdiA family of two-partner secretion proteins. CdiA effectors carry diverse C-terminal toxin domains (CdiA-CT), which are delivered into neighboring target cells to inhibit growth. CDI+ bacteria also produce CdiI immunity proteins that bind specifically to cognate CdiA-CT toxins and protect the cell from auto-inhibition. Here, we compare the structures of homologous CdiA-CT/CdiI complexes from Escherichia coli EC869 and Yersinia pseudotuberculosis YPIII to explore the evolution of CDI toxin/immunity protein interactions. Both complexes share an unusual β-augmentation interaction, in which the toxin domain extends a β-hairpin into the immunity protein to complete a six-stranded anti-parallel sheet. However, the specific contacts differ substantially between the two complexes. The EC869 β-hairpin interacts mainly through direct H-bond and ion-pair interactions, whereas the YPIII β-hairpin pocket contains more hydrophobic contacts and a network of bridging water molecules. In accord with these differences, we find that each CdiI protein only protects target bacteria from its cognate CdiA-CT toxin. The compact β-hairpin binding pocket within the immunity protein represents a tractable system for the rationale design of small molecules to block CdiA-CT/CdiI complex formation. We synthesized a macrocyclic peptide mimic of the β-hairpin from EC869 toxin and solved its structure in complex with cognate immunity protein. These latter studies suggest that small molecules could potentially be used to disrupt CDI toxin/immunity complexes.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Robert P. Morse, Julia L.E. Willett, Parker M. Johnson, Jing Zheng, Alfredo Credali, Angelina Iniguez, James S. Nowick, Christopher S. Hayes, Celia W. Goulding
Contact-dependent growth inhibition (CDI) is a widespread mechanism of inter-bacterial competition mediated by the CdiB/CdiA family of two-partner secretion proteins. CdiA effectors carry diverse C-terminal toxin domains (CdiA-CT), which are delivered into neighboring target cells to inhibit growth. CDI+ bacteria also produce CdiI immunity proteins that bind specifically to cognate CdiA-CT toxins and protect the cell from auto-inhibition. Here, we compare the structures of homologous CdiA-CT/CdiI complexes from Escherichia coli EC869 and Yersinia pseudotuberculosis YPIII to explore the evolution of CDI toxin/immunity protein interactions. Both complexes share an unusual β-augmentation interaction, in which the toxin domain extends a β-hairpin into the immunity protein to complete a six-stranded anti-parallel sheet. However, the specific contacts differ substantially between the two complexes. The EC869 β-hairpin interacts mainly through direct H-bond and ion-pair interactions, whereas the YPIII β-hairpin pocket contains more hydrophobic contacts and a network of bridging water molecules. In accord with these differences, we find that each CdiI protein only protects target bacteria from its cognate CdiA-CT toxin. The compact β-hairpin binding pocket within the immunity protein represents a tractable system for the rationale design of small molecules to block CdiA-CT/CdiI complex formation. We synthesized a macrocyclic peptide mimic of the β-hairpin from EC869 toxin and solved its structure in complex with cognate immunity protein. These latter studies suggest that small molecules could potentially be used to disrupt CDI toxin/immunity complexes.
Graphical abstract
Categories: Journal Articles
Bacterial Networks in Cells and Communities
Publication date: 20 November 2015
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Victor Sourjik, Julia A. Vorholt
Research on the bacterial regulatory networks is currently experiencing a true revival, driven by advances in methodology and by emergence of novel concepts. The biannual conference Bacterial Networks (BacNet15) held in May 2015, in Sant Feliu de Guíxols, Spain, covered progress in the studies of regulatory networks that control bacterial physiology, cell biology, stress responses, metabolism, collective behavior and evolution. It demonstrated how interdisciplinary approaches that combine molecular biology and biochemistry with the latest microscopy developments, whole cell (-omics) approaches and mathematical modeling can help understand design principles relevant in microbiology. It further showed how current biotechnology and medical microbiology could profit from our knowledge of and ability to engineer regulatory networks of bacteria.
Graphical abstract
Source:Journal of Molecular Biology, Volume 427, Issue 23
Author(s): Victor Sourjik, Julia A. Vorholt
Research on the bacterial regulatory networks is currently experiencing a true revival, driven by advances in methodology and by emergence of novel concepts. The biannual conference Bacterial Networks (BacNet15) held in May 2015, in Sant Feliu de Guíxols, Spain, covered progress in the studies of regulatory networks that control bacterial physiology, cell biology, stress responses, metabolism, collective behavior and evolution. It demonstrated how interdisciplinary approaches that combine molecular biology and biochemistry with the latest microscopy developments, whole cell (-omics) approaches and mathematical modeling can help understand design principles relevant in microbiology. It further showed how current biotechnology and medical microbiology could profit from our knowledge of and ability to engineer regulatory networks of bacteria.
Graphical abstract
Categories: Journal Articles
Protein Structure is Related to RNA Structural Reactivity in vivo
Publication date: Available online 17 November 2015
Source:Journal of Molecular Biology
Author(s): Yin Tang, Sarah M. Assmann, Philip C. Bevilacqua
We assessed whether in vivo mRNA structural reactivity and the structure of the encoded protein are related. This is the first investigation of such a relationship that utilizes information on RNA structure obtained in living cells. Based on our recent genome-wide Structure-seq analysis in Arabidopsis thaliana, we report that, as a meta property, regions of individual mRNAs that code for protein domains generally have higher reactivity to dimethyl sulfate (DMS), a chemical that covalently modifies accessible As and Cs, than regions that encode protein domain junctions. This relationship is prominent for proteins annotated for catalytic activity and reversed in proteins annotated for binding and transcription regulatory activity. Upon analyzing intrinsically disordered proteins, we found a similar pattern for disordered regions as compared to ordered regions: regions of individual mRNAs that code for ordered regions have significantly higher DMS reactivity than regions that code for intrinsically disordered regions. Based on these effects, we hypothesize that the decreased DMS reactivity of RNA regions that encode protein domain junctions or intrinsically disordered regions may reflect increased RNA structure which may slow translation, allowing time for the nascent protein domain or ordered region of the protein to fold, thereby reducing protein misfolding. In addition, a drop in DMS reactivity was observed on portions of mRNA sequences that correspond to the C-termini of protein domains, suggesting ribosome protection at these mRNA regions. Structural relationships between mRNAs and their encoded proteins may have evolved to allow efficient and accurate protein folding.
Graphical abstract
Source:Journal of Molecular Biology
Author(s): Yin Tang, Sarah M. Assmann, Philip C. Bevilacqua
We assessed whether in vivo mRNA structural reactivity and the structure of the encoded protein are related. This is the first investigation of such a relationship that utilizes information on RNA structure obtained in living cells. Based on our recent genome-wide Structure-seq analysis in Arabidopsis thaliana, we report that, as a meta property, regions of individual mRNAs that code for protein domains generally have higher reactivity to dimethyl sulfate (DMS), a chemical that covalently modifies accessible As and Cs, than regions that encode protein domain junctions. This relationship is prominent for proteins annotated for catalytic activity and reversed in proteins annotated for binding and transcription regulatory activity. Upon analyzing intrinsically disordered proteins, we found a similar pattern for disordered regions as compared to ordered regions: regions of individual mRNAs that code for ordered regions have significantly higher DMS reactivity than regions that code for intrinsically disordered regions. Based on these effects, we hypothesize that the decreased DMS reactivity of RNA regions that encode protein domain junctions or intrinsically disordered regions may reflect increased RNA structure which may slow translation, allowing time for the nascent protein domain or ordered region of the protein to fold, thereby reducing protein misfolding. In addition, a drop in DMS reactivity was observed on portions of mRNA sequences that correspond to the C-termini of protein domains, suggesting ribosome protection at these mRNA regions. Structural relationships between mRNAs and their encoded proteins may have evolved to allow efficient and accurate protein folding.
Graphical abstract
Categories: Journal Articles
Progress and current challenges in modeling large RNAs
Publication date: Available online 14 November 2015
Source:Journal of Molecular Biology
Author(s): Srinivas Somarowthu
Recent breakthroughs in next-generation sequencing technologies have led to the discovery of several classes of non-coding RNAs (ncRNAs). It is now apparent that RNA molecules are not just carriers of genetic information, but are also key players in many cellular processes. While there has been a rapid increase in the number of ncRNA sequences deposited in various databases over the past decade, the biological functions of these ncRNAs are largely not well understood. Similar to proteins, RNA molecules carry out a function by forming specific three-dimensional structures. Understanding the function of a particular RNA therefore requires a detailed knowledge of its structure. However, determining experimental structures of RNA is extremely challenging. In fact, RNA-only structures represent just 1% of the total structures deposited in the PDB. Thus, computational methods that predict three-dimensional RNA structures are in high demand. Computational models can provide valuable insights into structure-function relationships in ncRNAs, and can aid in the development of functional hypotheses and experimental designs. In recent years, a set of diverse RNA structure prediction tools have become available, which differ in computational time, input data and accuracy. This review discusses the recent progress and challenges in RNA structure prediction methods.
Graphical abstract
Source:Journal of Molecular Biology
Author(s): Srinivas Somarowthu
Recent breakthroughs in next-generation sequencing technologies have led to the discovery of several classes of non-coding RNAs (ncRNAs). It is now apparent that RNA molecules are not just carriers of genetic information, but are also key players in many cellular processes. While there has been a rapid increase in the number of ncRNA sequences deposited in various databases over the past decade, the biological functions of these ncRNAs are largely not well understood. Similar to proteins, RNA molecules carry out a function by forming specific three-dimensional structures. Understanding the function of a particular RNA therefore requires a detailed knowledge of its structure. However, determining experimental structures of RNA is extremely challenging. In fact, RNA-only structures represent just 1% of the total structures deposited in the PDB. Thus, computational methods that predict three-dimensional RNA structures are in high demand. Computational models can provide valuable insights into structure-function relationships in ncRNAs, and can aid in the development of functional hypotheses and experimental designs. In recent years, a set of diverse RNA structure prediction tools have become available, which differ in computational time, input data and accuracy. This review discusses the recent progress and challenges in RNA structure prediction methods.
Graphical abstract
Categories: Journal Articles
HER2 Transmembrane Domain Dimerization Coupled with Self-Association of Membrane-Embedded Cytoplasmic Juxtamembrane Regions
Publication date: Available online 14 November 2015
Source:Journal of Molecular Biology
Author(s): Pavel E. Bragin, Konstantin S. Mineev, Olga V. Bocharova, Pavel E. Volynsky, Eduard V. Bocharov, Alexander S. Arseniev
Receptor tyrosine kinases of the human epidermal growth factor receptor (HER or ErbB) family transduce biochemical signals across plasma membrane, playing a significant role in vital cellular processes and in various cancers. Inactive HER/ErbB receptors exist in equilibrium between the monomeric and unspecified pre-dimerized states. After ligand binding, the receptors are involved in strong lateral dimerization with proper assembly of their extracellular ligand-binding, single-span transmembrane, and cytoplasmic kinase domains. The dimeric conformation of the HER2 transmembrane domain that is believed to support the cytoplasmic kinase domain configuration corresponding to the receptor active state was previously described in lipid bicelles. Here we used high-resolution NMR spectroscopy in another membrane-mimicking micellar environment and identified an alternative HER2 transmembrane domain dimerization coupled with self-association of membrane-embedded cytoplasmic juxtamembrane region. Such a dimerization mode appears to be capable of effectively inhibiting the receptor kinase activity. This finding refines the molecular mechanism regarding the signal propagation steps from the extracellular to cytoplasmic domains of HER/ErbB receptors.
Graphical abstract
Source:Journal of Molecular Biology
Author(s): Pavel E. Bragin, Konstantin S. Mineev, Olga V. Bocharova, Pavel E. Volynsky, Eduard V. Bocharov, Alexander S. Arseniev
Receptor tyrosine kinases of the human epidermal growth factor receptor (HER or ErbB) family transduce biochemical signals across plasma membrane, playing a significant role in vital cellular processes and in various cancers. Inactive HER/ErbB receptors exist in equilibrium between the monomeric and unspecified pre-dimerized states. After ligand binding, the receptors are involved in strong lateral dimerization with proper assembly of their extracellular ligand-binding, single-span transmembrane, and cytoplasmic kinase domains. The dimeric conformation of the HER2 transmembrane domain that is believed to support the cytoplasmic kinase domain configuration corresponding to the receptor active state was previously described in lipid bicelles. Here we used high-resolution NMR spectroscopy in another membrane-mimicking micellar environment and identified an alternative HER2 transmembrane domain dimerization coupled with self-association of membrane-embedded cytoplasmic juxtamembrane region. Such a dimerization mode appears to be capable of effectively inhibiting the receptor kinase activity. This finding refines the molecular mechanism regarding the signal propagation steps from the extracellular to cytoplasmic domains of HER/ErbB receptors.
Graphical abstract
Categories: Journal Articles